溫馨提示×
您好,登錄后才能下訂單哦!
點擊 登錄注冊 即表示同意《億速云用戶服務條款》
您好,登錄后才能下訂單哦!
這期內容當中小編將會給大家帶來有關DECoN中最高分辨率的CNV檢測工具怎么用,文章內容豐富且以專業的角度為大家分析和敘述,閱讀完這篇文章希望大家可以有所收獲。
DECoN是一款CNV檢測工具,適用于exon-based的panel測序,可以識別single exon CNV
panel測序在臨床上應用廣泛,目前利用panel測序數據來檢測SNP是比較成熟的,而CNV的檢測則缺乏有效的工具。在這樣的背景下,DECoN應運而生,開發者在ExomeDepth軟件的的基礎上進一步修改,主要有以下兩點大的改動
新增了檢測染色體上第一個外顯子區域的變異
在隱馬可夫模型中新增了exon之間的距離這一因素
通過模擬數據和真實數據對軟件的性能進行評估,在模擬數據集中,DECoN效果驚人,100%的靈敏度和99%的特異性。真實數據采用了illumina TruSight Cancer Panel測序的結果,最終鑒定出來24個exon CNV,用MLPA技術進行驗證,有23個可以檢測到,假陽性率4%,更加詳細的評估結果請查看文章中的描述。
該軟件的運行速度也非常快,還提供了良好的結果可視化,示意如下
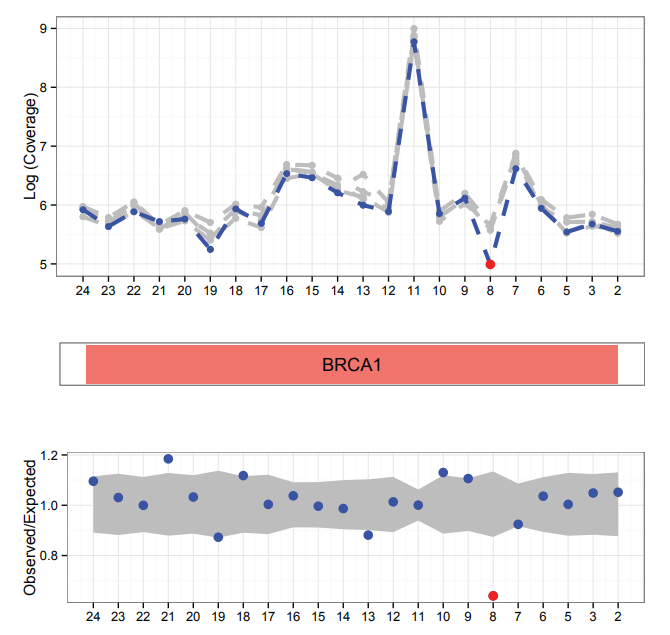
上面的折線圖展示的是基因上coverage的分布,灰色代表對照樣本,藍色代表實驗樣本;中間展示的是基因的名稱,最下方的散點圖代表觀測值和期望值之間的比值,灰色區域代表95%置信區間,當比值顯著偏離置信區間時,認為該區域存在拷貝數變異。上圖所示的紅點區域代表實際觀測值小于期望值,說明發生了deletion。
軟件的源代碼保存在github上,鏈接如下
https://github.com/RahmanTeam/DECoN
具體操作分為以下4步, 對應4個R腳本
讀取bam文件,計算coverage, 用法如下
Rscript ReadInBams.R \
--bams bamList.txt \
--bed Target_Regions.bed \
--fasta hg19.fa \
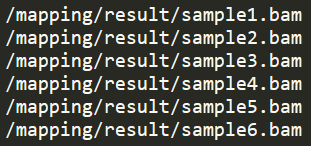
--out DECoNtest輸入文件為bam文件的列表,目的區域的bed文件,參考基因組的fasta文件,bam文件的格式如下
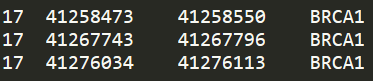
目的區域bed文件的格式如下
輸出結果是一個后綴為RData的文件,保存了樣本的coverage信息,該軟件中用FPKM值來表示。
進行質量控制,檢測coverage過度的exon區域,相關性較差的樣本等,用法如下
Rscript IdentifyFailures.R \
--Rdata DECoNtest.RData \
--exons customNumbering.txt \
--mincorr .98 \
--mincov 100 \
--custom TRUE \
--out DECoNtest輸入文件為第一步產生的RData文件,另外還需要自定義的exon編號的文件
customNumbering.txt
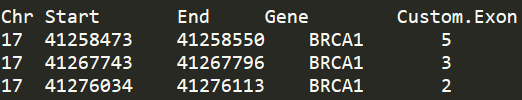
內容示意如下
如果所有的樣本和exon區域都符合要求,則該命令不會輸出結果,如果有不合格的樣本和區域,則需要剔除之后在進行操作。
進行CNV calling,用法如下
Rscript makeCNVcalls.R \
--Rdata DECoNtest.RData \
--exons customNumbering.txt \
--custom TRUE \
--out DECoNtestCalls \
--plot All \
–-plotFolder DECoNTestPlots通過R包Shiny構建了一個基于瀏覽器的交互式結果展示頁面,用法如下
Rscript runShiny.R \
--Rdata DECoNtestCalls.RData可以查看coverage分布圖,cnv calling的結果等信息,示意如下
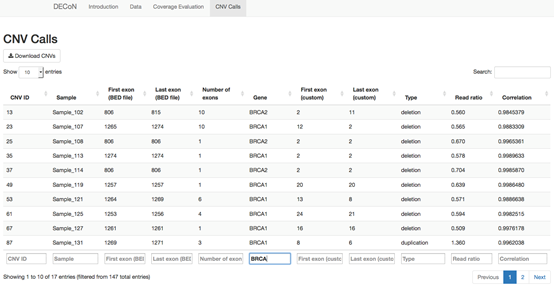
對于panel測序的CNV檢測,推薦使用DECoN進行分析。
上述就是小編為大家分享的DECoN中最高分辨率的CNV檢測工具怎么用了,如果剛好有類似的疑惑,不妨參照上述分析進行理解。如果想知道更多相關知識,歡迎關注億速云行業資訊頻道。
免責聲明:本站發布的內容(圖片、視頻和文字)以原創、轉載和分享為主,文章觀點不代表本網站立場,如果涉及侵權請聯系站長郵箱:is@yisu.com進行舉報,并提供相關證據,一經查實,將立刻刪除涉嫌侵權內容。





