溫馨提示×
您好,登錄后才能下訂單哦!
點擊 登錄注冊 即表示同意《億速云用戶服務條款》
您好,登錄后才能下訂單哦!
這篇文章主要介紹“如何使用R包SomaticSignatures進行denovo的signature推斷”,在日常操作中,相信很多人在如何使用R包SomaticSignatures進行denovo的signature推斷問題上存在疑惑,小編查閱了各式資料,整理出簡單好用的操作方法,希望對大家解答”如何使用R包SomaticSignatures進行denovo的signature推斷”的疑惑有所幫助!接下來,請跟著小編一起來學習吧!
原文在:http://bioconductor.org/packages/release/bioc/vignettes/SomaticSignatures/inst/doc/SomaticSignatures-vignette.html
library(SomaticSignatures)
library(SomaticCancerAlterations)
library(BSgenome.Hsapiens.1000genomes.hs37d5)
sca_metadata = scaMetadata()
sca_metadata
sca_data = unlist(scaLoadDatasets())
sca_data$study = factor(gsub("(.*)_(.*)", "\\1", toupper(names(sca_data))))
sca_data = unname(subset(sca_data, Variant_Type %in% "SNP"))
sca_data = keepSeqlevels(sca_data, hsAutosomes(), pruning.mode = "coarse")
sca_vr = VRanges(
seqnames = seqnames(sca_data),
ranges = ranges(sca_data),
ref = sca_data$Reference_Allele,
alt = sca_data$Tumor_Seq_Allele2,
sampleNames = sca_data$Patient_ID,
seqinfo = seqinfo(sca_data),
study = sca_data$study)
sca_vr
可以看到,這個包,需要的是sca_data這個變量里面各個列,用到了的就是 c( "Sample","chr", "pos","ref", "alt") 這些列。所以我們自己的somatic突變信息,也需要制作成為這5列。
在文章主頁下載;https://static-content.springer.com/esm/art%3A10.1038%2Fs41422-020-0333-6/MediaObjects/41422_2020_333_MOESM23_ESM.csv
這個是大于500M的CSV文件,下載后修改名字,然后讀入R,并且制作成為 SomaticSignatures 包的輸入數據的代碼如下:
library(data.table)
b=fread('../maf.csv',data.table = F)
b[1:4,1:3]
colnames(b)
mut=b
table(mut$Variant_Type)
mut=mut[mut$Variant_Type=='SNP',]
a=mut[,c(10,2,3,8,9)]
colnames(a)=c( "Sample","chr", "pos","ref", "alt")
alls=as.character(unique( a$Sample))
a$study=a$Sample
head(a)
雖然我們使用了 data.table 包的 fread函數,可以超級快的讀入大于500M的CSV文件,但是也需要一點時間啦。
制作的a這個變量,如下:
> head(a)
Sample chr pos ref alt study
2 FP1705100059DN01 chr1 4870770 G T FP1705100059DN01
3 FP1705100059DN01 chr1 5111686 C T FP1705100059DN01
4 FP1705100059DN01 chr1 5116099 C T FP1705100059DN01
5 FP1705100059DN01 chr1 5151401 C T FP1705100059DN01
6 FP1705100059DN01 chr1 5151403 G C FP1705100059DN01
7 FP1705100059DN01 chr1 5217189 G A FP1705100059DN01
一個很普通的數據框而已,并不是SomaticSignatures 包的文檔介紹sca_data這個變量的類型,但是該有的5列信息是有的。
sca_vr = VRanges(
seqnames = a$chr ,
ranges = IRanges(start = a$pos,end = a$pos+1),
ref = a$ref,
alt = a$alt,
sampleNames = as.character(a$Sample),
study=as.character(a$study))
sca_vr
在SomaticSignatures 包已經是封裝好的函數,很容易就可以獲取,而且速度超級快哦,代碼如下:
# 突變位點坐標基于hg19,從基因組根據坐標獲取堿基上下文
sca_motifs = mutationContext(sca_vr, BSgenome.Hsapiens.UCSC.hg19)
head(sca_motifs)
# 對每個樣本,計算 96 突變可能性的 比例分布情況
escc_sca_mm = motifMatrix(sca_motifs, group = "study", normalize = TRUE)
dim( escc_sca_mm )
table(colSums(escc_sca_mm))
head(escc_sca_mm[,1:4])
我們都知道,sanger研究所科學家【1】提出來了腫瘤somatic突變的signature概念 ,把96突變頻譜的非負矩陣分解后的30個特征,在cosmic數據庫可以學習它。不同的特征有不同的生物學含義【2】,比如文章【3】 就是使用了 這些signature區分生存!主要是R包deconstructSigs可以把自己的96突變頻譜對應到cosmic數據庫的30個突變特征。
但是我們現在要自己推斷denovo的signature,所以使用SomaticSignatures 包的identifySignatures函數哦,代碼如下:
# 預先設定待探索的 signature 數量范圍,文章最后選定11個
if(F){
n_sigs = 5:15
gof_nmf = assessNumberSignatures(escc_sca_mm , n_sigs, nReplicates = 5)
save(gof_nmf,file = 'gof_nmf.Rdata')
}
load(file = 'gof_nmf.Rdata')
# 這個 assessNumberSignatures 步驟耗時很嚴重。
plotNumberSignatures(gof_nmf)
# 根據這個圖表,選擇11個 signature
sigs_nmf = identifySignatures(escc_sca_mm ,
11, nmfDecomposition)
save(escc_sca_mm,sigs_nmf,file = 'escc_denovo_results.Rata')
代碼如下:
load(file = 'escc_denovo_results.Rata')
str(sigs_nmf)
library(ggplot2)
plotSignatureMap(sigs_nmf) + ggtitle("Somatic Signatures: NMF - Heatmap")
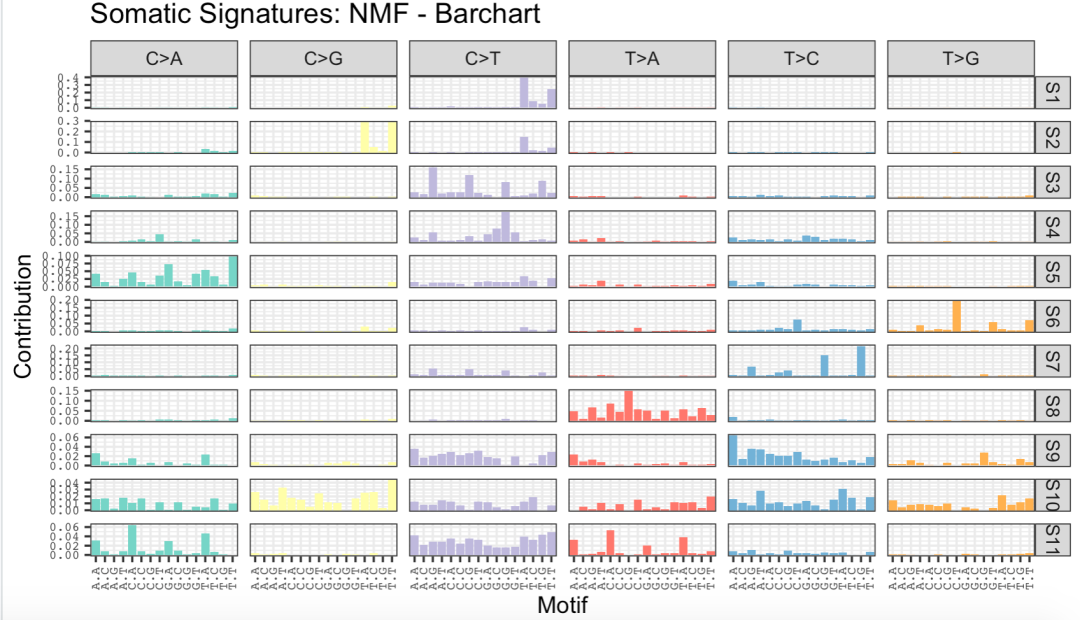
plotSignatures(sigs_nmf, normalize =T) +
ggtitle("Somatic Signatures: NMF - Barchart") +
facet_grid(signature ~ alteration,scales = "free_y")
出圖如下:
到此,關于“如何使用R包SomaticSignatures進行denovo的signature推斷”的學習就結束了,希望能夠解決大家的疑惑。理論與實踐的搭配能更好的幫助大家學習,快去試試吧!若想繼續學習更多相關知識,請繼續關注億速云網站,小編會繼續努力為大家帶來更多實用的文章!
免責聲明:本站發布的內容(圖片、視頻和文字)以原創、轉載和分享為主,文章觀點不代表本網站立場,如果涉及侵權請聯系站長郵箱:is@yisu.com進行舉報,并提供相關證據,一經查實,將立刻刪除涉嫌侵權內容。





