溫馨提示×
您好,登錄后才能下訂單哦!
點擊 登錄注冊 即表示同意《億速云用戶服務條款》
您好,登錄后才能下訂單哦!
這篇文章主要介紹“如何使用HiCUP進行Hi-C數據預處理”,在日常操作中,相信很多人在如何使用HiCUP進行Hi-C數據預處理問題上存在疑惑,小編查閱了各式資料,整理出簡單好用的操作方法,希望對大家解答”如何使用HiCUP進行Hi-C數據預處理”的疑惑有所幫助!接下來,請跟著小編一起來學習吧!
HiCUP是一款經典的Hi-C數據預處理軟件,官網如下
https://www.bioinformatics.babraham.ac.uk/projects/hicup/
數據處理的流程示意如下
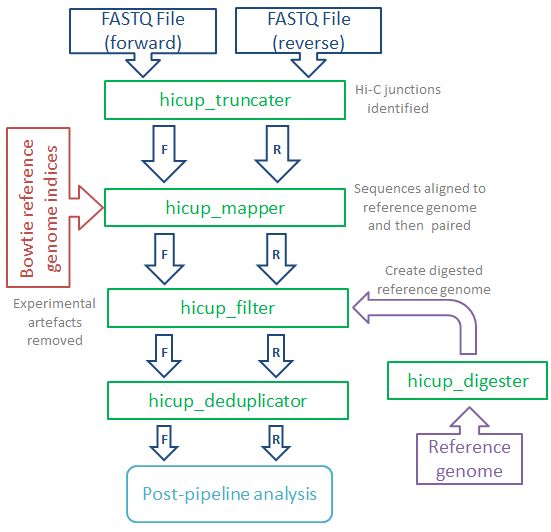
首先通過hicup_truncater識別原始序列中的junction reads, 最典型的Hi-C的reads如下所示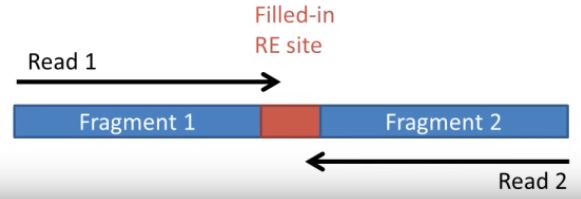
R1和R2來自兩個不同的fragments, 當然這取決于插入偏度長度和讀長的關系,當連接點與fragment兩端的距離小于測序讀長是,會發生下圖所示的情況
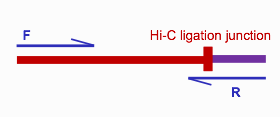
其中一端的序列是一個嵌合體序列,這樣的序列在后續比對時會被過濾掉。為了保留這部分有效reads,hicup_truncater根據酶切位點的特征來識別所有reads上的連接位點,從而識別上圖中的嵌合體序列,并對這樣的序列末端進行切割,切除多余的嵌合體序列。切割完之后,這樣的序列和普通的R1,R2就一樣了,可以進行后續的mapping。
hicup_mapper將雙端reads與參考基因組比對,由于Hi-C文庫的R1和R2來源于空間結構近的染色質,其線性距離比傳統的雙端測序插入片段的長度大的多,如果直接進行雙端比對,覺得部分reads都比對不上參考基因組,所以這里是對每一端的序列分別比對,然后再進行合并。
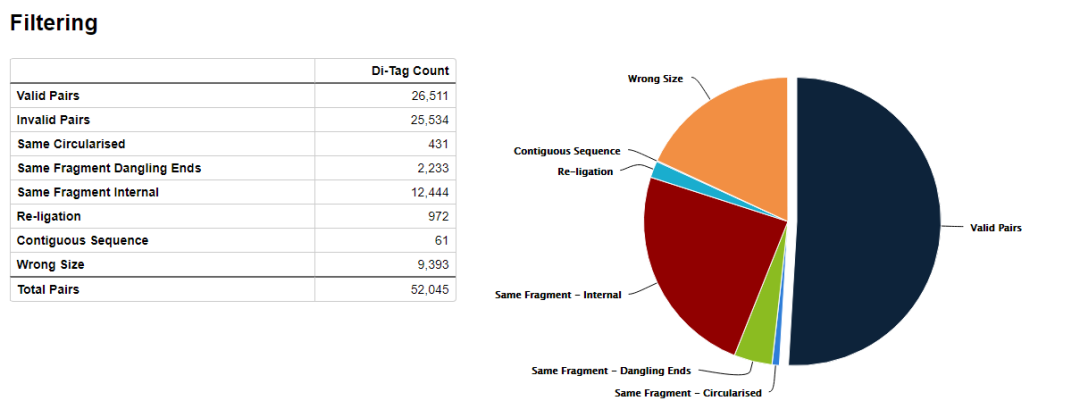
hicup_filter對比對上的序列進行過濾,如下圖所示
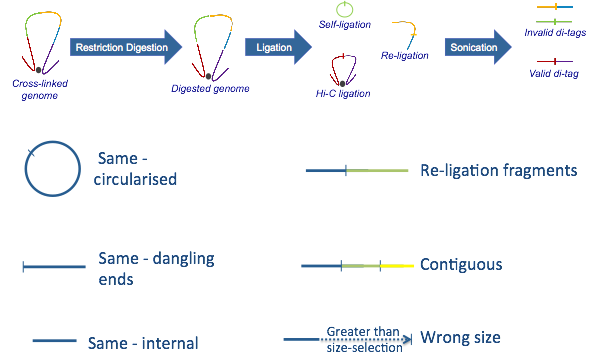
只保留valid di-tags, 其他諸如selft-ligation, Re-ligation等片段都會被過濾掉。
hicup_deduplicator用來去除PCR重復,因為valid reads的多少用來表征染色質互作的頻率,PCR重復的reads數量會對這個信息造成干擾,如果不去除PCR重復,junction reads的數目多可能是PCR重復多,不一定是因為染色質交互頻率強而導致的reads多。
軟件的安裝也很方便,直接下載解壓縮即可。使用步驟如下
所有的參考基因組比對軟件都需要事先對基因組建立索引,HiCUP支持使用bowtie或bowtie2進行比對,以bowtie2為例,建立基因組索引的方式如下
bowite2-build hg19.fa hg19
第一個參數是基因組的fasta文件,第二個參數是輸出的索引文件的名稱。
采用hicup_digester這個腳本來創建基因組的酶切圖譜,基本用法如下
hicup_digester \
--re1 A^AGCTT,HindIII \
--genome hg19_digester_db \
hg19.fa根據限制性內切酶識別的位點,將基因組序列進行模擬酶切,得到所有可能的酶切片段。--re1指定切割位點的序列和內切酶的名字,--genome指定輸出文件的名稱。最終輸出的文件名示例如下
Digest_hg19_digester_db_HindIII_None_09-46-07_17-05-2019.txt
首先通過如下命令生成一個配置文件的模板
hicup --example
該命令會生成一個名為hicup_example.conf的文件,在此基礎上進行編輯就可以了。在配置中對每個選項都體用了詳細的注釋,根據需求修改即可。常用的修改的選項如下
#Path to the reference genome indices
#Remember to include the basename of the genome indices
Index: /bi/scratch/Genomes/Human/GRCh48/Homo_sapiens.GRCh48
#Path to the genome digest file produced by hicup_digester
Digest: /bi/scratch/Genomes/Human/GRCh48/Digest_Homo_sapiens_GRCh48_HindIII_None_14-43-31_10-02-2016.txt.gz
#FASTQ files to be analysed, placing paired files on adjacent lines
s_1_1_sequence.fastq.gz
s_1_2_sequence.fastq.gz包括基因組索引和酶切圖譜的路徑,以及需要處理的Hi-C原始fastq文件的路徑。
準備好配置文件之后,就可以運行了,用法如下
hicup --config hicup.conf
在輸出結果的目錄會生成一個html文件,展示了質控的各項指標,內容如下所示
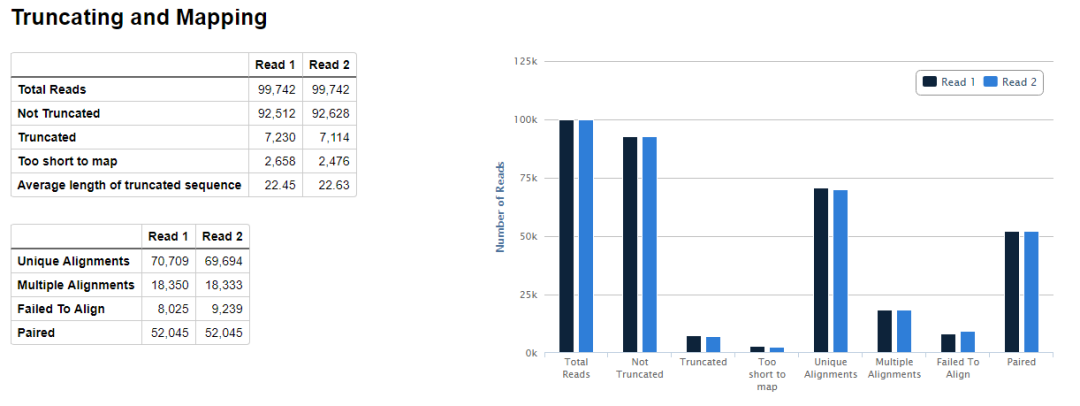
示意如下,可以看到valid pairs的比例在50%左右
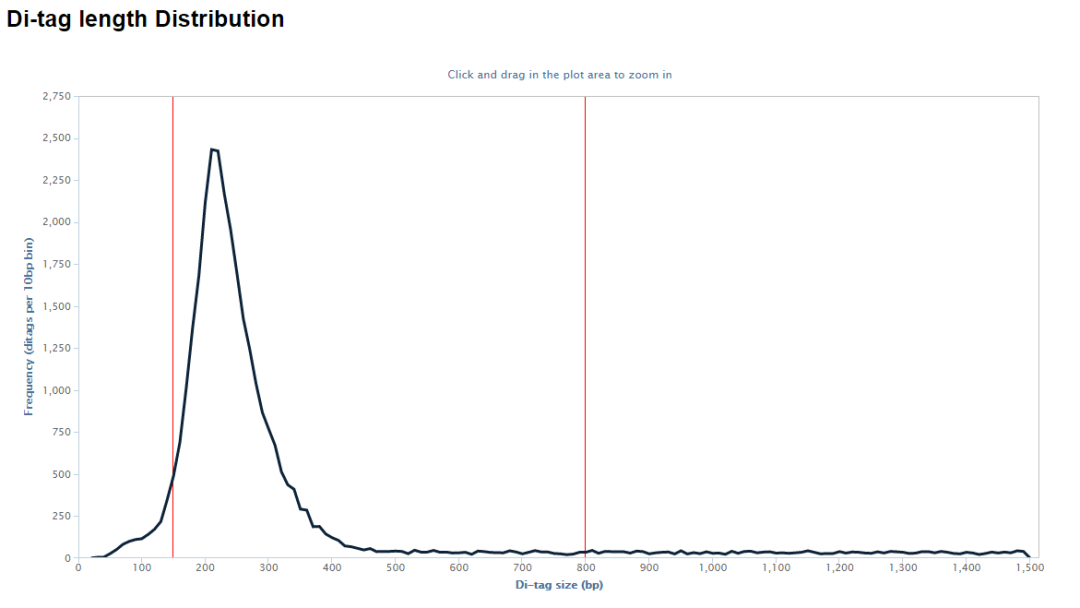
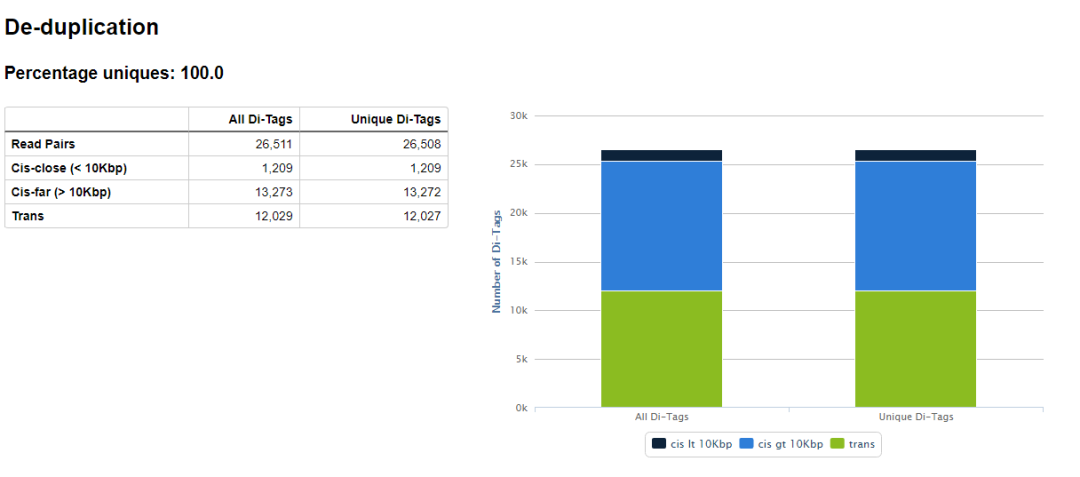
除此之外,輸出目錄還有很多的文件,其中后綴為hicup_bam的文件包含了最終的de-duplication之后的reads的比對結果,可以用于下游的分析。
到此,關于“如何使用HiCUP進行Hi-C數據預處理”的學習就結束了,希望能夠解決大家的疑惑。理論與實踐的搭配能更好的幫助大家學習,快去試試吧!若想繼續學習更多相關知識,請繼續關注億速云網站,小編會繼續努力為大家帶來更多實用的文章!
免責聲明:本站發布的內容(圖片、視頻和文字)以原創、轉載和分享為主,文章觀點不代表本網站立場,如果涉及侵權請聯系站長郵箱:is@yisu.com進行舉報,并提供相關證據,一經查實,將立刻刪除涉嫌侵權內容。





