溫馨提示×
您好,登錄后才能下訂單哦!
點擊 登錄注冊 即表示同意《億速云用戶服務條款》
您好,登錄后才能下訂單哦!
這期內容當中小編將會給大家帶來有關怎么使用tophat-fusion鑒定融合基因,文章內容豐富且以專業的角度為大家分析和敘述,閱讀完這篇文章希望大家可以有所收獲。
tophat-fusion 是一款利用RNA_seq 數據鑒定融合基因的工具。
該軟件是集成在tophat軟件中的,只需要安裝好tophat之后就可以使用了,使用方法也比較簡單,唯一需要注意的是目錄結構。
tophat-fusion要求固定的目錄結構,比如我在result文件夾下進行tophat-fusion的分析, 那么我需要在該目錄下準備以下幾個文件
物種對應的refGene.txt 和 ensGene.txt, 這兩個文件可以從UCSC下載得到
新建一個blast 文件夾,注意文件夾的名字必須為”blast”, 在blast 文件夾下需要從NCBI下載nt, human_genomic, other_genomic開頭的所有文件,下載的鏈接如下:
ftp://ftp.ncbi.nlm.nih.gov/blast/db/
結果輸出目錄,每個樣本對應一個輸出目錄,輸出目錄的前綴為tophat_, 下劃線之后加上樣本名稱,類似tophat_MCF,MCF為樣本的名字
當然你還需要物種對應的bowtie1的索引文件,注意這里必須為bowtie1的索引, tophat檢測融合基因時推薦bowtie1的索引方式。
上述文件都準備好之后,就可以開始分析了,步驟如下
第一步其實就是利用tophat將reads比對到參考基因組上,只不過對于融合基因的reads而言,其比對方式比較特殊,需要添加額外的參數,具體代碼如下
tophat2 -o tophat_MCF7 -p 20 --fusion-search --keep-fasta-order --bowtie1 --no-coverage-search -r 0 --mate-std-dev 80 --max-intron-length 100000 --fusion-min-dist 100000 --fusion-anchor-length 13 --fusion-ignore-chromosomes chrM hg19_bowtie1/hg19 SRR064286_1.fastq SRR064286_2.fastq
在result目錄下,直接運行如下代碼就可以了
tophat-fusion-post -p 20 --num-fusion-reads 1 --num-fusion-pairs 2 --num-fusion-both 5 hg19_bowtie1/hg19
默認處理的是human的融合基因,如果是其他物種,需要添加--non-human參數。
tophat-fusion會根據目錄結構自動識別對應的樣本, 運行完成之后,會生成一個名為tophatfusion_out的文件夾,該文件夾下是所有樣本的融合基因分析結果。
我們只需要看其中的result.html文件就可以了,內容示意如下
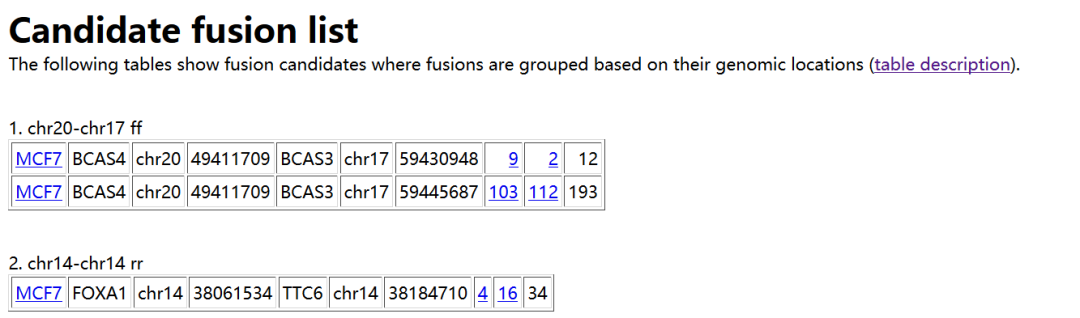
每一列的含義如下
Sample name in which a fusion is identified
Gene on the “left” side of the fusion
Chromosome ID on the left
Coordinates on the left
Gene on the “right” side
Chromosome ID on the right
Coordinates on the right
Number of spanning reads
Number of spanning mate pairs
Number of spanning mate pairs where one end spans a fusion
相比fusionmap, 該軟件的運行時間特別的長。
上述就是小編為大家分享的怎么使用tophat-fusion鑒定融合基因了,如果剛好有類似的疑惑,不妨參照上述分析進行理解。如果想知道更多相關知識,歡迎關注億速云行業資訊頻道。
免責聲明:本站發布的內容(圖片、視頻和文字)以原創、轉載和分享為主,文章觀點不代表本網站立場,如果涉及侵權請聯系站長郵箱:is@yisu.com進行舉報,并提供相關證據,一經查實,將立刻刪除涉嫌侵權內容。





