溫馨提示×
您好,登錄后才能下訂單哦!
點擊 登錄注冊 即表示同意《億速云用戶服務條款》
您好,登錄后才能下訂單哦!
今天給大家介紹一下HLA-VBSeq中如何對全基因組數據進行HLA分型。文章的內容小編覺得不錯,現在給大家分享一下,覺得有需要的朋友可以了解一下,希望對大家有所幫助,下面跟著小編的思路一起來閱讀吧。
HLA-VBseq 利用全基因組測序的數據,可以提供8位的HLA分型結果,其文獻鏈接如下
https://bmcgenomics.biomedcentral.com/articles/10.1186/1471-2164-16-S2-S7
下面利用30X的全基因組數據,對HLA-VBSeq, PHLAT, HLAminer這3款軟件的分型結果進行了評估,準確率匯總如下
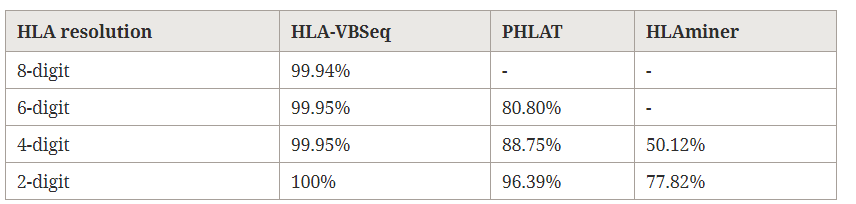
可以看到,只有HLA-VBSeq提供了8位的分型結果,準確率高達99.94%;對于2位到4位的分型結果,其準確率也高于另外兩款軟件。
同時還評估了不同測序量時,各種軟件提供的4位分型結果的準確率,結果如下
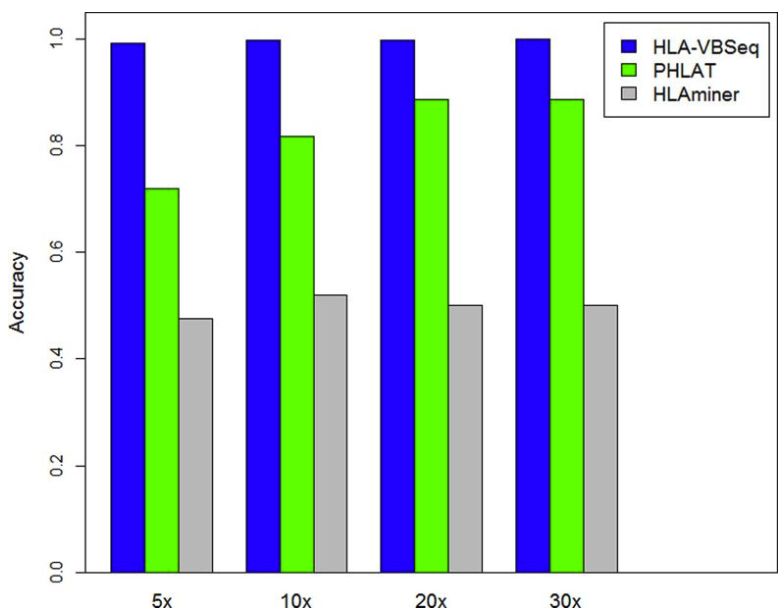
在不同條件下,HLA-VBseq的準確率都是最高的。由此可見,該軟件的分型效果還是相當不錯的,官網如下
http://nagasakilab.csml.org/hla/
該軟件采用java語言開發,直接下載HLAVBseq.jar就可以了,除了該文件之外,還需要下載以下幾個文件
bamNameIndex.jar
SamToFastq.jar
parse_result.pl
hla_all.fasta
Allelelist.txt
前三個程序在處理fastq文件時會用到;后兩個文件是從IMGA/HLA數據庫下載的,如果覺得官網提供的版本較老,可以從IMGA/HLA數據庫下載最新版。
軟件的步驟較多,首先將fastq序列與參考基因組進行比對,得到bam文件,然后對該bam文件進行操作。步驟如下:
利用samtools view 命令挑選出比對到HLA區域的reads , 命令如下
samtools view -hb align.bam chr6:29907037-29915661 chr6:31319649-31326989 chr6:31234526-31241863 chr6:32914391-32922899 chr6:32900406-32910847 chr6:32969960-32979389 chr6:32778540-32786825 chr6:33030346-33050555 chr6:33041703-33059473 chr6:32603183-32613429 chr6:32707163-32716664 chr6:32625241-32636466 chr6:32721875-32733330 chr6:32405619-32414826 chr6:32544547-32559613 chr6:32518778-32554154 chr6:32483154-32559613 chr6:30455183-30463982 chr6:29689117-29699106 chr6:29792756-29800899 chr6:29793613-29978954 chr6:29855105-29979733 chr6:29892236-29899009 chr6:30225339-30236728 chr6:31369356-31385092 chr6:31460658-31480901 chr6:29766192-29772202 chr6:32810986-32823755 chr6:32779544-32808599 chr6:29756731-29767588 | samtools fastq - -1 R1.fq -2 R2.fq
需要注意的是,在使用view命令時,雖然也可以直接提供一個bed格式的文件來挑選特定區域的reads,但是這種用法不會利用到bam文件的索引,所以速度很慢。對于全基因組數據,bam文件很大,上述寫法雖然冗長,但是執行效率高。
利用samtools view 命令挑選出沒有比對上參考基因組的reads, 命令如下:
samtools view -hb -f 12 /home/pub/output/WGS/18B0315D/6343/6343_final.bam | samtools fastq - -1 unmapped_R1.fq -2 unmapped_R2.fq
將比對到HLA區域的reads和沒比對上參考基因組的reads合并,命令如下
cat R1.fq unmapped_R1.fq > R1.fastq cat R2.fq unmapped_R2.fq > R2.fastq
利用bwa軟件,將上一步得到的reads與HLA參考序列比對,命令如下
bwa index hla_all.fasta bwa mem -t 8 -P -L 10000 -a hla_all.fasta R1.fastq R2.fastq > out.sam
HLA-VBSeq支持雙端或者單端測序的數據,這里以雙端數據為例,用法如下
java -jar HLAVBSeq.jar hla_all.fasta out.sam result.txt --alpha_zero 0.01 --is_paired
上一步就已經生成結果了,這一步只是格式化,下面的代碼會篩選出HLA-A基因的分型結果
perl parse_result.pl Allelelist.txt result.txt | grep "^A\*" | sort -k2 -n -r > HLA.txt
格式化之后的結果,內容如下
A*01:01:01:01 17.4022266628604 A*11:01:01 12.0376819868684
共兩列,第一列為Allel, 第二列為該Allel區域的平均測序深度。
以上就是HLA-VBSeq中如何對全基因組數據進行HLA分型的全部內容了,更多與HLA-VBSeq中如何對全基因組數據進行HLA分型相關的內容可以搜索億速云之前的文章或者瀏覽下面的文章進行學習哈!相信小編會給大家增添更多知識,希望大家能夠支持一下億速云!
免責聲明:本站發布的內容(圖片、視頻和文字)以原創、轉載和分享為主,文章觀點不代表本網站立場,如果涉及侵權請聯系站長郵箱:is@yisu.com進行舉報,并提供相關證據,一經查實,將立刻刪除涉嫌侵權內容。





