溫馨提示×
您好,登錄后才能下訂單哦!
點擊 登錄注冊 即表示同意《億速云用戶服務條款》
您好,登錄后才能下訂單哦!
這篇文章主要介紹stringTie工具有什么用,文中介紹的非常詳細,具有一定的參考價值,感興趣的小伙伴們一定要看完!
對于轉錄組數據而言,最基礎的分析就是基因和轉錄本水平的定量了,定量就是確定一個基因或者轉錄本的表達量,其中定量的方式有很多種。
最直接的方式就是統計mapping到這個基因/轉錄本上的reads的個數,將reads數作為表達量。我們稱這種表達量為raw count。
在raw count的基礎上,利用外顯子長度進行歸一化,就得到了TPM值的定量方式。對于每個基因,將raw count除了該基因的長度(exon長度之和) , 得到長度歸一化之后的表達量。某個基因的TPM值就是利用歸一化之后的表達量,計算了一個相對豐度。具體計算公式如下,注意基因長度以k為單位

在raw count的基礎上,利用測序量和外顯子長度兩個因素進行歸一化,就得到了RPKM/FPKM值的定量方式。首先將raw count除了mapping 上的所有reads數,得到相對豐度,在除以該基因長度(exon長度之和), 就可以計算出RPKM值。測試時每一條插入片段稱為一個fragment, 對于雙端測序,一個fragment 會得到兩條reads。
RPKM和FPKM 唯一不同的地方在于raw count的計算,RPKM 計算的是reads 數,而FPKM 值計算的是fragments 數,對于單端測序, fragment 和 reads 的個數是相等的;對于雙端測序,reads 數目是fragments 數目的兩倍,對于FPKM 而言,即使雙端的兩條reads都比對上了基因組,在計數時也知計一次,因為兩條reads來源于同一個fragment。
具體計算公式如下, 需要注意單位,mapping上的reads 總數以M為單位,基因長度以k為單位。
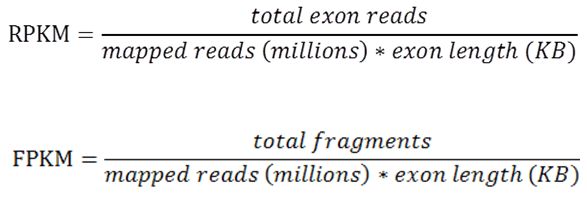
能夠進行定量的軟件有很多,本文主要介紹stringTie這款軟件。
在早期的轉錄組數據分析中,最經典的分析策略是tophat+cufflinks+cuffdiff, 這套分析的pipeline會給出基于FPKM值的定量結果,然后進行差異分析,但是隨著測序數據量的提高和分析手段的發展,這套分析策略出現了很多的問題。
首先就是tophat的速度很慢,相比新出的比對軟件,其速度可以算得上是龜速了,同樣的數據量,hisat/star只需要半個小時就可以比對完成,tophat2至少需要5到6個小時;其次,基于FPKM值得到的差異結果和實驗手段如qPCR驗證的一致性較差。
為了順應測序和分析的新趨勢,原本的開發團隊對整個pipeline進行了全面升級, 用hisat 代替tophat, 用stringTie + ballgown 代替cufflinks + cuffdiff。
stringTie 可以看做是cufflinks 軟件的升級版本,其功能和cufflinks是一樣的 ,包括下面兩個主要功能
轉錄本組裝
定量
相比cuffinks, 其運行速度更快。該軟件的官網如下
https://ccb.jhu.edu/software/stringtie/index.shtml
stringTie的輸入文件為經過排序之后的bam文件,常見用法有以下幾種
對于模式生物,如human, mouse等,通常只需要對已知的轉錄本定量即可,用法如下
stringtie -p 10 \ -G hg19.gtf \ -o output.gtf \ -b ballgown_out_dir -e \ align.sorted.bam
-G參數指定參考基因組的gtf文件,-o指定輸出的文件,格式也為gtf, -b指定ballgown的輸出結果目錄,這個參數是為了方便下游進行ballgown差異分析,-e參數要求軟件只輸出已知轉錄本的定量結果。
在輸出的GTF格式的文件中,對于每個轉錄本,會給出以下3種表達量
coverage
TPM
FPKM
對于單個樣本進行組裝,用法如下
stringtie align.sorted.bam -o assembly.gtf -p 20 -G hg19.gtf
在組裝的轉錄本中,也會給出定量的結果,對于組裝的新轉錄本和基因,默認采用STRG加數字編號進行區分,示例如下
gene_id "STRG.1" transcript_id "STRG.1.1"
單個樣本組裝完成后,會合并所有樣本的轉錄本組裝結果,得到一個非冗余的轉錄本集合,用法如下
stringtie --merge \ -o assembly.gtf \ -p 20 \ -G hg19.gtf \ sampleA.gtf sampleB.gtf
在合并的非冗余轉錄本中,采用MSTRG加數字編號對基因和轉錄本進行編號,示例如下
gene_id "MSTRG.2" transcript_id "MSTRG.2.2"
本質上,stringTie只提供了轉錄本水平的表達量,定量方式包括TPM和FPKM值兩種。為了進行raw count的定量方式,官方提供了prepED.py腳本,可以計算出raw count的表達量,用法如下
python prepDE.py \ -i sample_list.txt \ -g gene_count_matrix.csv \ -o transcript_count_matrix.csv
輸入文件為sample_list.txt, 該文件為\t分隔的兩列,第一列為樣本名稱,第二列為定量的gtf文件的路徑,示例如下
sampleA A.stringtie.gtf sampleB B.stringtie.gtf
同時輸出基因和轉錄本水平的raw count表達量值。
采用stringTie進行定量,運行速度快是一個優勢,同時提供raw count, FPKM, TPM 3種定量方式的結果,也是其最便利的地方。
以上是“stringTie工具有什么用”這篇文章的所有內容,感謝各位的閱讀!希望分享的內容對大家有幫助,更多相關知識,歡迎關注億速云行業資訊頻道!
免責聲明:本站發布的內容(圖片、視頻和文字)以原創、轉載和分享為主,文章觀點不代表本網站立場,如果涉及侵權請聯系站長郵箱:is@yisu.com進行舉報,并提供相關證據,一經查實,將立刻刪除涉嫌侵權內容。





